Interprétation d’un résultat de NGS
Le NGS (Next-Generation Sequencing) ou séquençage à haut débit est une technique d’analyse qui permet, notamment en oncologie, d’étudier simultanément un grand nombre de gènes, voire le génome complet, et d’identifier ainsi des anomalies moléculaires oncogéniques, comme des mutations sur l’ADN tumoral et/ou des gènes de fusions sur l’ARN tumoral, dans le but d’établir le profil moléculaire d’une tumeur pour pouvoir proposer un traitement adapté. Cette technique permet à la fois l’analyse des biomarqueurs connus et des biomarqueurs émergents 2.
Prélèvements et informations nécessaires
Tissu tumoral, prélèvement cytologique ou biopsie liquide sont des prélèvements compatibles à la réalisation d’une NGS, avec une sensibilité de la technique plus élevée pour les échantillons tissulaires.
Différentes informations doivent être connues car elles peuvent impacter les résultats du NGS et leur interprétation :
- La date, la nature et la qualité du prélèvement ;
- Le contexte clinique et le moment du prélèvement puisqu’ils influencent le choix du panel analysé, les gènes d’intérêt étant différents d’un contexte clinique à un autre mais aussi, pour un même contexte, au diagnostic (recherche de biomarqueurs diagnostiques et/ou prédictifs) ou en cours de traitement (recherche de biomarqueurs de suivi de l’évolution de la pathologie ou de résistance à une thérapie ciblée) ;
- Le délai entre le prélèvement et l’analyse puisqu’il peut avoir un impact sur la qualité de l’échantillon extrait (ADN ou ARN) et sur la sensibilité de l’analyse ;
- La qualité de l’échantillon, notamment la cellularité tumorale (pourcentage de cellules tumorales de l’échantillon qui doit être supérieur à la limite de détection de la méthode utilisée) et le fixateur utilisé (certains fixateurs, comme le liquide de Bouin, sont à proscrire) ;
- Le panel utilisé avec la liste des gènes analysés.
Quelques définitions 3
- Les reads sont les séquences des fragments d’ADN et d’ARN lues au cours du NGS ;
- La profondeur correspond au nombre de lecture de chaque base, exprimée en X. Une profondeur minimale à atteindre est définie comme un seuil de qualité ;
- La couverture représente le pourcentage du panel pour lequel la profondeur de séquençage est supérieure à la profondeur minimale prédéfinie ;
- La sensibilité est la probabilité qu’un dispositif donne un résultat positif en présence du marqueur cible.
Quelles informations sur les mutations ?
Pour chaque mutation identifiée, le compte-rendu doit préciser 4 :
- Le gène concerné,
- Le type de mutation retrouvée (substitution, délétion, insertion),
- Sa localisation,
- Son impact sur la protéine correspondante.
L’utilisation d’une nomenclature des mutations somatiques, par exemple celle proposée par Escande et al 4,est fortement recommandée par l’INCa (Institut National du Cancer) (tableau) [3,4].
Dans le compte-rendu, tous les variants recensés doivent être décrits précisément selon la nomenclature HGVS avec le nom du gène, la séquence génomique de référence (code NM_), la description de la mutation et de l’altération protéique correspondante [Figure 1].
Le NGS permet d’évaluer la fréquence mutationnelle, c’est à dire le pourcentage d’allèles mutés dans l’échantillon analysé. Une fréquence mutationnelle inférieure à la sensibilité indiquée pour la technique utilisée suggère la possibilité d’un faux positif.
Les altérations moléculaires identifiées par le NGS doivent être hiérarchisées selon leur caractère oncogénique et le fait qu’il s’agisse ou non de mutations actionnables avec une thérapie ciblée en regard.
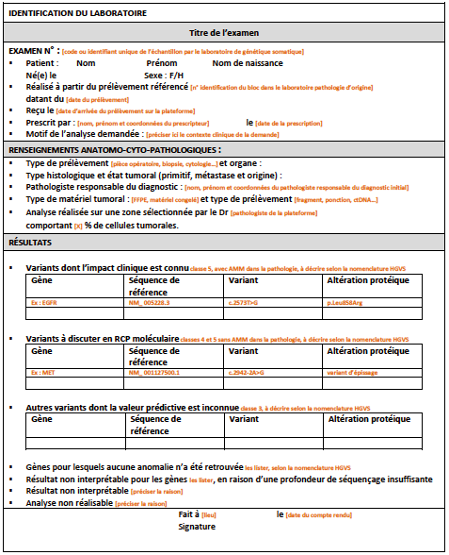
[Figure 1]
Exemple de compte-rendu de génétique
Conclusion du compte-rendu
Outre les données concernant la méthode utilisée, la conclusion du compte-rendu de NGS doit répertorier et décrire précisément les altérations moléculaires identifiées 5. Celles-ci seront discutées ensuite en RCP (Réunion de Concertation Pluridisciplinaire) d’oncologie ou de génétique moléculaire, facilitant l’accès à certaines thérapies ciblées et contribuant également à identifier les patients éligibles à l’inclusion dans des essais cliniques guidés par la génomique.
(1) D’après Denicolaï E, Tomasini P. La Lettre du Cancérologue 2020 ; 9 : 585-6
(2) Kalemkerian GP, Narula N, Kennedy EB et al. Molecular Testing Guideline for the Selection of Patients With Lung Cancer for Treatment With Targeted Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice Guideline Update. J Clin Oncol. 2018;36(9):911-919
(3) Groupe de travail INCa. Séquençage de nouvelle génération d’un panel de gènes pour l’analyse en génétique somatique/Validation de la méthode (2016). www.e-cancer.fr
(4) Escande F, Rouleau E. Nomenclature des variations de séquence en génétique : application en génétique somatique. Correspondances en Onco-Théranostic 2015 ;4 :119-24
(5) INCa. Modèle de compte-rendu de génétique moléculaire pour la recherche de mutations somatiques dans les tumeurs solides. www.e-cancer.fr